Understanding battleground earth
8 Nov 2018 by Evoluted New Media








Falk Hildebrand takes through the first global study of the soil microbiome. This incredible international effort uncovered worrying human impact, antibiotic production and one hell of a microbial battle. You’ll never look at a patch of ground the same way again…
Soil is full of life, something that is essential for nutrient cycling and carbon storage.
To better understand how it functions, my colleague, Professor Mohammad Bahram from the University of Uppsala and I designed the first global study of bacteria and fungi in soil, which we conducted with an international research team.
Our results – published in Nature1 – show that bacteria and fungi are in constant competition for nutrients and produce an arsenal of antibiotics to gain an advantage over one another. The study can help predict the impact of climate change on soil, and help us make better use of natural soil components in agriculture.
The soil microbiome is composed of thousands of species. So far, it is the most diverse microbial environment known to us, surpassing the usual gastrointestinal diversity by several orders of magnitude (although the gastrointestinal system has a higher density of microorganisms), while the oceans diversity and density in microorganisms is notably lower.
In the soil there is a lively mix of different organisms, going in scale from annelids and insects to microorganisms like fungi, bacteria and archaea, which represent the largest fraction in soil in terms of numbers. The soil microbiome is not only of immense importance to keep our lawns and forests healthy, it is of utmost importance for our high-yield farming industry. This is because it is a vital part of global nutrient recycling. For example, the nitrogen cycle is key to plant growth and is based on a symbiotic relationship between plants and fungi/bacteria in the rhizosphere: the soil surrounding plant roots. Understanding and modelling the nutrient recycling is therefore of direct importance for the agricultural industry.
Together with Professor Bahram, we designed a study to research the soil microbiome on a global scale, describing fungal, bacterial and nutrient distributions, to understand associations between these and identify patterns that hold across the globe .
Microbe metagenomics Over the course of five years, our international collaboration collected 58.000 soil samples from 1450 sites over all continents (40 subsamples per site, see also L. Tedersoo et al. 20142). These sites were carefully selected to be unaffected by human activities such as agriculture. Of the 1450 sites sampled, we selected 189 for in-depth analysis (Figure 1), covering the world’s most important biomes, from tropical forests to tundra. We used high-throughput methods to simultaneously describe the phylogeny, function and abundance of microbes in these samples. We could not use classical microbiology methods, such as culturing, restriction fragment length polymorphism or microscopy, as these had never been designed for high-throughput research. To circumvent this restriction, we extensively used metagenomic and amplicon sequencing – sequencing based methods developed in the last decade. Amplicon sequencing (also referred to as metabarcoding or metataxonomics) is the targeted PCR amplification of phylogenetically informative genes and their subsequent sequencing. In this study we decided to amplify three different genomic regions, in order to amplify the DNA of different kingdoms. First, we used a 16S rDNA gene region to cover bacterial diversity, second the 18S rDNA gene to target microeukaryotes and third the ITS (internal transcribed spacer) region between the 18S and 28S gene with primers specific to fungi.
[caption id="attachment_68729" align="alignnone" width="620"] Figure 1. The 189 sample sites spread over 5 continents (colored dots) + 145 local soil samples (black dots).[/caption]
Figure 1. The 189 sample sites spread over 5 continents (colored dots) + 145 local soil samples (black dots).[/caption]
Regional differences The distribution of bacteria and fungi showed regional differences. Most striking was that bacteria are largely found everywhere, having the highest genetic diversity in temperate zones with a moderate climate. They seem to follow the hypothesis “Everything is everywhere, but the environment selects”4.
Environmental factors have the most influence on bacterial abundance: we identified pH as one of the driving factors and bacteria seem to prefer hot and wet locations. Fungi are usually more prevalent in colder and dryer climates like the tundra, which we interpret as fungi (maybe aided by their hyphae, the long cellular outgrowth allowing for long distance transport of nutrients) being able to cope with dryness better. Importantly, they are much less restricted by environmental factors than bacteria, but instead tend to be more geographically restricted (Figure 2). However, the ratio of C/N (a proxy for nutrient availability) was also very important for fungal composition.
Simply put: one can expect the fungi found in two close sites to be very similar, while for bacteria it is more important that soil pH, inorganic compound concentration and humidity are comparable if the same bacteria are to be discovered. Thus, our data suggests that there is a clear niche differentiation between fungi and bacteria in the soil. This also implies specialists for different environments and considerable competition for nutrients. And where there is competition, there is usually conflict.
[caption id="attachment_68730" align="alignnone" width="620"]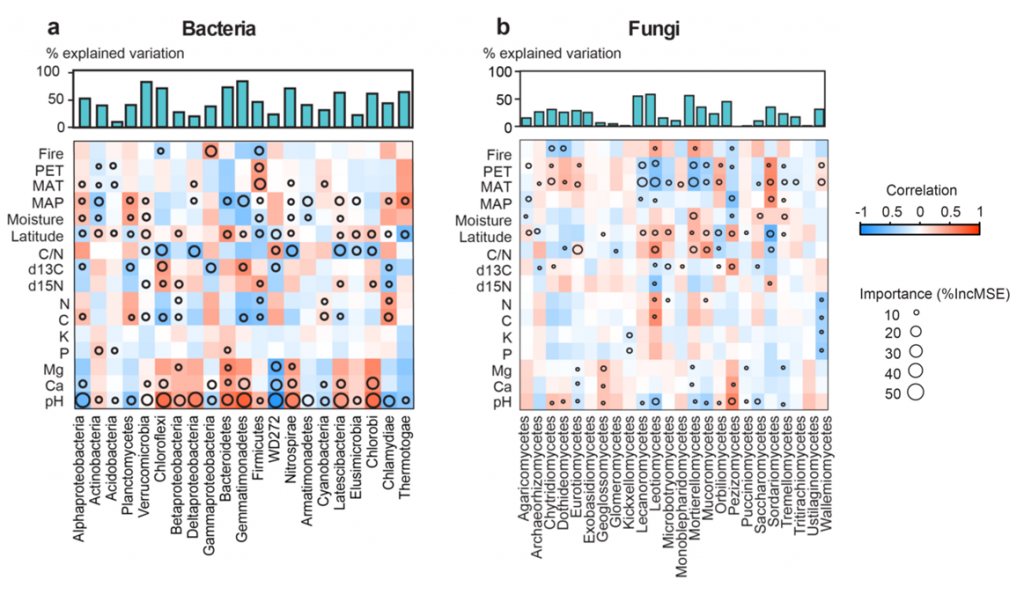 Figure 2: The main bacterial (a) and fungal (b) taxa and their correlation to environmental variables. Note that bacteria are in general stronger correlated to these than fungi, and that the distribution suggests niche differentiation between the two groups. pH, soil pH representing soil pH and its quadratic term; ?15N, nitrogen stable isotope signature; ?13C, carbon stable isotope signature; PET, potential of evapotranspiration; Fire, time from the last fire disturbance; NPP, net primary productivity.[/caption]
Figure 2: The main bacterial (a) and fungal (b) taxa and their correlation to environmental variables. Note that bacteria are in general stronger correlated to these than fungi, and that the distribution suggests niche differentiation between the two groups. pH, soil pH representing soil pH and its quadratic term; ?15N, nitrogen stable isotope signature; ?13C, carbon stable isotope signature; PET, potential of evapotranspiration; Fire, time from the last fire disturbance; NPP, net primary productivity.[/caption]
Global microbial war One of the key, but puzzling, observations in our soil samples is that bacterial diversity is lower if the relative proportion of fungi is higher. This coincides with a “phylogenetic thinning out” for bacteria, indicative of a purging process: eliminating organisms that do not carry a specific adaptation. We would expect a very strong selection to be responsible for such a clear signal, like for example antibiotics. Hence, we investigated antibiotic resistance in these samples. This showed a very strong link between the number of antibiotic resistance genes (ARG) in bacteria and the proportion of fungi in a given soil site, especially those with potential for antibiotics production such as Penicillium.
This pattern could well be explained by the fact that fungi produce antibiotics in warfare with bacteria, and only bacteria with adequate ARG can survive this. This hypothesis was further supported by multiple lines of independent evidence. For example, fungal, but not bacteria, biomass was positively correlated to the fraction of bacterial ARG and we also found putative fungal genes for producing antibiotics to correlate strongly to the proportion of bacterial ARG. This antagonism between fungi and bacteria influences the overall diversity of bacterial communities and determines their genetic repertoire of antibiotic resistance. Now this information in turn can be used to predict the spread of genes that lead to antibiotic resistance in different ecosystems, and via what routes they may reach human pathogens. It could also help predict and pinpoint locations with high levels of natural antibiotics producers. And in more detail, predict in which environments to expect especially severe interkingdom competition, and therefore especially effective antibiotics – and resistances to these.
Effects of human activity When comparing data from the unspoiled soil sites with data from locations affected by humans, such as farmland or garden lawns, the ratios between bacteria, fungi and antibiotics were completely different. We think that this shift in the natural balance – that probably evolved over most of the earth’s history – shows the effect of human activities on the soil microbiome, with unknown consequences so far. However, a better understanding of the interactions between fungi and bacteria in soil could help to reduce the usage of soil fertilizer in agriculture, as one could give beneficial microorganisms a better chance at survival in their natural environment. The observed correlation between ARGs and the ratio of fungi to bacteria was very strong in our global soil dataset, but does it also hold in other soil samples and other environments? For this we tested publicly available metagenomes from soil sites. While this showed the same key fungi (Ascomycota, Basidiomycota) being correlated to bacterial ARG, these trends were weaker. This illustrates a common problem of working with public samples: different laboratories have different protocols for soil collection, DNA extraction, sample storage and sequencing platform, all leading to study specific biases that are hard to control for on heterogenous public data. Hence, we also tested the ARG trend on an additional 145 samples collected by us with our protocols on a local scale (Figure 1). On this data we could perfectly replicate trends from the global dataset, showing that this is a general trend in soils.
But what about other environments? Testing the large scale Tara oceans5 sampling efforts showed that in ocean samples other fungal taxa dominated, with the fungi-like protist Oomycota being highly abundant. Again, for the latter group we found a strong correlation to ocean bacterial ARGs, which was surprising since Oomycota are mostly known to be parasites. Last, we also calculated correlations in human gut and oral samples. Here, no significant correlation of the bacterial/fungal ratio to ARG could be detected. This is interesting as it could mean different things: first it could mean that the human microbiome is already disturbed due to the constant low level exposure from our food and environment, largely discussed in literature as “hygiene hypothesis”6. Second, it is well known that most metagenomic studies do contain precious little non-bacterial DNA. This is, simply put, mostly a problem with the DNA extraction protocols being optimized towards breaking open bacterial cells, and even this is inconsistent between protocols7. The advancement of metagenomics allows the exploration of microbial communities at a massive scale. In this study, we describe the relative contributions of bacteria and fungi to nutrient cycling that are different around the world. We can infer that global climate change may affect their composition and function in soil differently. Tipping this balance might also change the fraction of fungi competing against bacteria with antibiotics.
References:
- Bahram, M., Hildebrand, F., et al. Structure and function of the global topsoil microbiome. Nature, published online 1 August 2018. DOI: 10.1038/s41586-018-0386-6
- Tedersoo, L. et al. 2014. “Global Diversity and Geography of Soil Fungi.” Science 346(6213): 1256688–1256688. http://www.sciencemag.org/cgi/doi/10.1126/science.1256688 (November 28, 2014).
- Tedersoo, Leho et al. 2015. “Shotgun Metagenomes and Multiple Primer Pair-Barcode Combinations of Amplicons Reveal Biases in Metabarcoding Analyses of Fungi.” MycoKeys 10: 1–43. http://mycokeys.pensoft.net/articles.php?id=4852 (May 14, 2015).
- Baas-Becking, L. G. M. 1934. Geobiologie; Of Inleiding Tot de Milieukunde. W. P. Van Stockum & Zoon NV, The Hague.
- Sunagawa, Shinichi et al. 2015. “Structure and Function of the Global Ocean Microbiome.” Science 348(6237): 1261359–1261359. http://www.sciencemag.org/content/348/6237/1261359.abstract (May 23, 2015).
- Strachan, D P. 2000. “Family Size, Infection and Atopy: The First Decade of the ‘Hygiene Hypothesis’.” Thorax 55 Suppl 1: S2-10. http://www.ncbi.nlm.nih.gov/pubmed/10943631.
- Costea, Paul I. et al. 2017. “Towards Standards for Human Fecal Sample Processing in Metagenomic Studies.” Nature Biotechnology 35(11): 1069–76. http://dx.doi.org/10.1038/nbt.3960.

Author:
Falk Hildebrand is postdoctoral fellow at the European Molecular Biology Laboratory (EMBL)
