Getting through the data maze
8 Sep 2009 by Evoluted New Media
Scientists working in the areas of gene and protein research are being increasingly overwhelmed by the vast amounts of data being produced, says Carl-Johan Ivarsson. Can computer software offer a way to tame this problem of information overload and allow scientists to analyse the data themselves?
Scientists working in the areas of gene and protein research are being increasingly overwhelmed by the vast amounts of data being produced, says Carl-Johan Ivarsson. Can computer software offer a way to tame this problem of information overload and allow scientists to analyse the data themselves?
 GENE expression profiling is an important way of studying a wide variety of diseases such as breast cancer and leukaemia. Broadly speaking, gene expression profiling refers to the measurement of the activity (the expression) of tens of thousands of genes at once, to create a global picture of cellular function. These profiles can then, for example, be used to distinguish between cells that are actively dividing, or to show how the cells react to a particular treatment.
GENE expression profiling is an important way of studying a wide variety of diseases such as breast cancer and leukaemia. Broadly speaking, gene expression profiling refers to the measurement of the activity (the expression) of tens of thousands of genes at once, to create a global picture of cellular function. These profiles can then, for example, be used to distinguish between cells that are actively dividing, or to show how the cells react to a particular treatment.
The problem facing researchers working in this specific area of molecular biology, however, lies in the vast amount of data that is created by gene expression experiments. With such a large volume of data to consider, it is impossible to derive any real biological meaning from these findings with the naked eye alone, which means that sophisticated data algorithms need to be developed in order for researchers to interpret the data effectively.
For scientists working in this field, this presents a serious problem; in order for current research projects to yield the most useful results, it is absolutely vital to capture, explore, and analyse this kind of data effectively so that researchers can consider sub-groups (such as patients who are in remission versus patients who have suffered a relapse), whilst also examining the different types of cell abnormalities related to each condition.
As such, gene expression profiling offers a good example of how the scientist (biologist, MD, and so on) can begin to regain control of the analysis, when the amount of data is large.
Up until now, most software programs have focused on being able to handle increasingly vast amounts of data, which means that the role of the scientist/researcher has been largely set aside. As a result, a lot of data analysis is now performed by bioinformaticians and biostatisticians. However, in most cases, this model has several drawbacks, since it is typically the scientist who knows the most about biology. Fortunately, the latest technological advances in this area are making it much easier for scientists themselves to compare the enormous quantity of data generated, to test different hypothesis, and to explore alternative scenarios within seconds.
Not only that, but the latest bioinformatics software now enables scientists to analyse very large data sets by a combination of statistical methods and visualisation techniques such as Heatmaps and Principal Component Analysis (PCA). With the benefit of instant user feedback on all actions, as well as an intuitive user interface that can present all data in 3D, scientists studying diseases like childhood leukaemia can now easily analyse all of this important information in real-time, directly on their computer screen.
For all of these reasons, the latest computer software is increasingly playing a key role in unveiling important new discoveries, as it allows the actual researchers involved – the people with the most biological insight – to study the data and to look for patterns and structures, without having to be a statistics or computer expert.
Modern data analysis software can enable researchers to analyse and explore extremely large data sets – even those with more than 100 million data samples – on a regular PC. This kind of specialist software can even take advantage of annotations and other links that are connected with the data being studied, as well as a number statistical functions such as false discovery rates (FDR) and p-values.
"When you are looking at such a large amount of genetic data, there is bound to be a certain amount of 'technical and biological noise'," says Dr Anna Andersson, a researcher currently studying childhood leukaemia in the USA. "When you are presented with 30,000 small dots representing different expression levels on the screen, for example, many of them will be irrelevant to what you are studying, and so the data normalisation and filtering made possible by computer software plays a key role in making biological sense of the data."
For this reason, the latest data analysis software makes it very easy to select and test different levels of filtering, with features like one-click filter selection. Also, since researchers are provided with instant feedback of the filter results, it is very quick to find the right level of filtering required, making it much faster for them to find discriminating variables (genes), subgroups and patterns.
"With the help of this kind of data analysis software, you can test a hypothesis from the moment that you develop it," Dr Andersson says. "Testing different hypothesis used to be difficult, as earlier applications in this area were not nearly as flexible as they are today. In fact, we would often have to quit out of the application, re-load the data, and then test our new hypothesis. We are under time pressure like everyone else, and so the result is that you only test the most important and most likely hypotheses. Now, however, we tend to be much more creative with our theories, as we can easily test any number of hypotheses in rapid succession."
Less than 10 years ago, researchers were only able to work with analysis methods that provided information about single genes. The number of information points per subject has grown to hundreds of thousands in recent years, however, thanks to important technological advances in this area.
Most recently, perhaps within the past two years, the overall performance of data analysis software has been optimised significantly. In fact, with key actions and plots now displayed within a fraction of a second, researchers can increasingly perform the research they want and find the results they need instantly – without the wait.
For example, one of the methods used by Qlucore's software to visualise data is dynamic PCA, an innovative way of combining traditional PCA analysis with immediate user interaction. This software is able to manipulate the different PCA-plots – interactively and in real time – directly on the computer screen, and to work with all annotations and other links in a integrated way, all at the same time.
This approach has helped to open up new ways of working with the analysis and, as a consequence, has brought the biologists back into the analysis phase.
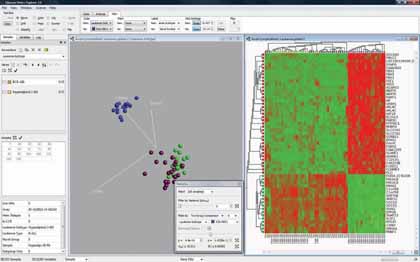 |
| Figure 1: "Software can be used to highlight the variables that discriminate the blue group from the rest" |
According to Dr Anderson, when performing her own research, she typically begins her analysis by opening up a dataset, and then taking a look at the inherent structures that form, without performing any filtering. Usually, if there are any outliers or natural groups forming, Dr Andersson can see it here. Next, she colours the samples based on their class, and then takes another look at the data to see if samples belonging to the same class form natural clusters.
Once this step is completed, Dr Andersson begins to filter the data by variance in order to reduce the noise levels. Usually, if natural classes were seen during her initial inspection, they will begin to form even tighter clusters when genes with no or very little variation are removed.
Finally, Dr Andersson uses the multigroup comparison (F-test based on Anova) to identify genes that significantly correlate with the classes. Once that process has been completed, she can export a variable (gene) list and a data matrix that will allow her to investigate the genes even further.
The process is similar when trying to identify unknown subtypes, except that here Dr Andersson uses the software to create a new class before marking the samples of the potential new class, and then cross-validates them before again exporting a variable (gene) list.
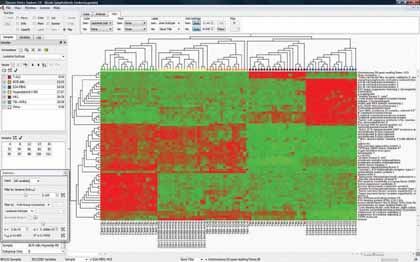 |
| Figure 2: "Plot types such as heatmap (pictured here) and Principal Component Analysis (PCA) are supported by software like Qlucore's Omics Explorer" |
"In our experience, when investigating similarities between healthy samples with that of our patient samples, it is very useful to first use the option of taking the normal samples, look at the structure forming in the principle component analysis, lock the principal components, and then to add the patient sample group," she explains. "Because the structure will change, however, a lot of applications will completely re-draw the findings, but applications like Qlucore's Omics Explorer now give us the option of locking the principle components, which makes the gene expression data much easier to interpret when comparing our patients' bone marrow samples with healthy samples, for example."
According to Andersson, the software works by projecting this high dimensional data down to lower dimensions, which can then be plotted in 3-dimentions on a computer screen and then rotated manually or automatically and examined by the naked eye. Different colours can make this analysis even easier, as each sub-group can be labelled with its own unique colour. As such, Dr Andersson can change her view of the data rapidly, so that she is only looking at the specific sub-group that interests her at any given moment.
In addition to this graphical data, 3D video can also be exported from the software easily. This is an especially important aspect of the software for researchers like Dr Andersson, since they typically seek to include interesting graphics in their presentations and related publications.
"With the freedom, speed and flexibility provided by modern data analysis software, it is possible to evaluate and test a number of different scenarios and hypothesis in a short time, and to fully understand the data being examined," Andersson adds. "This approach makes it possible for researchers to combine very large amounts of data, and therefore to conduct analysis in ways that were simply not possible before. After all, most researchers would agree: the larger the sample group, the better the statistics."
 |
| Figure 3: "With the right software, five synchronised plots can be viewed simultaneously" |
Although Dr Andersson is currently using Qlucore's software to study gene expression micro array data, other researchers have used it to study protein array data, miRNA data, and RT-PCR data as part of their research studies. The software has also been used to analyse protein data from 2-D gels, image analysis data, and in fact with any data set of multivariate data of sizes up to 1000 samples and 100,000 variables, or 1000 variables and 100,000 samples.
According to Dr Andersson, her own research efforts will continue to focus on understanding the molecular, genetic and chemical bases of leukemia in children.
"Of course, the ultimate dream is to find new and improved treatments that will be more effective against leukaemia, and which can target just the leukaemic cells, without harming any healthy cells. However, there are many things that we can do along the way while we continue to work towards that goal," Dr Andersson says. "With the data that we have now, we are starting to understand the genetic difference between different sub-groups, such as those with a better of worse prognosis, which means that we, in the future, might be able to administer treatments that are much more palatable for patients.
At the moment, even though the majority of the leukaemia patients are cured, some still succumb to the disease. If our research can help to pin-point such patients at the time of their diagnosis, and help identify which genetic networks are altered in these patients, I believe that is an important step forward."
