Mass spec goes 2D
19 Jan 2017 by Evoluted New Media



No longer side-lined as a computationally-hungry scientific curiosity, two-dimensional mass spectrometry is becoming a fully-fledged analytical technique says Maria van Agthoven
One of the cornerstones of biomedical research is proteomics, which studies protein expression in cells when they are under various conditions of stress, or how proteins respond to these conditions – for example, how proteins interact with pharmaceutical compounds.
Proteomes are very complex, and reverse phase liquid chromatography is often coupled with tandem mass spectrometry (LC-MS/MS) in order to maximise the amount of chemical information gathered from the analysis. In tandem mass spectrometry (MS/MS), ionized molecules (such as proteins or peptides) are fragmented in order to gain structural information (such as the peptide sequence and post-translational modifications). In order to properly correlate fragment ions with their precursors, a narrow range of mass-to-charge ratios is isolated before the fragmentation step.
Analytical scientists therefore have to make choices between co-eluting species in LC-MS/MS, which makes the analysis blind to low abundance compounds. Techniques like Precursor Acquisition Independent From Ion Count (PAcIFIC), developed by Professor David Goodlett at the University of Maryland, and SWATH, developed by SCIEX, attempt to bypass this problem by repeating LC runs while isolating ions over sliding mass-to-charge windows. But samples like whole cells or blood plasma have dynamic ranges that span over many orders of magnitude, and the development of sensitive analytical techniques which overcome the necessity of the isolation step in MS/MS is crucial for comprehensive biomedical research.
One mass spectrometer offers a more elegant solution to the problem of correlating precursor and fragment ions in tandem mass spectrometry: the Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR MS), which separates ions according to their mass-to-charge ratios by measuring their cyclotron frequencies in high magnetic fields. In the late 1980s, Tino Gäumann’s team in Switzerland took a leaf out of the NMR spectroscopy book and proposed a pulse sequence for an alternative method for tandem mass spectrometry they called two-dimensional mass spectrometry (2D MS). The maps obtained from this experiment showed the fragmentation patterns of all ions in a sample, without requiring any isolation beforehand. Unfortunately, the computational capacity needed to process and store the data was too high for 2D MS to be useful, and for almost two decades it was side-lined as a scientific curiosity.Analytical scientists therefore have to make choices between co-eluting species in LC-MS/MS, which makes the analysis blind to low abundance compounds
For the last seven years, teams from the University of Warwick in the United Kingdom (lead by Professor Peter O’Connor), the University of Lille (lead by Dr Christian Rolando), and the University of Strasbourg (lead by Dr Marc-André Delsuc) in France have revisited the topic of 2D MS on FT-ICR mass spectrometers in order to turn it into a fully-fledged analytical technique. We have optimised the pulse sequence to minimise the impact of artefacts, built an open access software package to process and visualise 2D mass spectra (Spectrometry Processing Innovative Kernel, or SPIKE), and developed algorithms to deal the scintillation noise problem inherent to any spectroscopy method with signal fluctuations.
We have applied 2D MS to peptides, small molecules, proteins, and tryptic digests of proteins of increasing complexity. Currently, we can acquire 2D mass spectra with laser-based fragmentation techniques and electron-based fragmentation techniques. With an IR laser, peptide ions fragment by losing post-translational modifications like phosphorylations or glycosylations before fragmenting along the peptidic chain, whereas with an electron-gun, peptide ions preferentially fragment along the peptidic chain. Acquiring 2D mass spectra with different fragmentation methods can therefore give us complementary structural information on proteins and peptides. The most complex sample we have published is the tryptic digest of collagen, a 400 kDa protein. We are currently in the process of developing algorithms to extract fragmentation patterns from 2D mass spectra in a way that allows us the interrogate proteomics databases like MASCOT or Prosight in order to automate data analysis of 2D mass spectra.
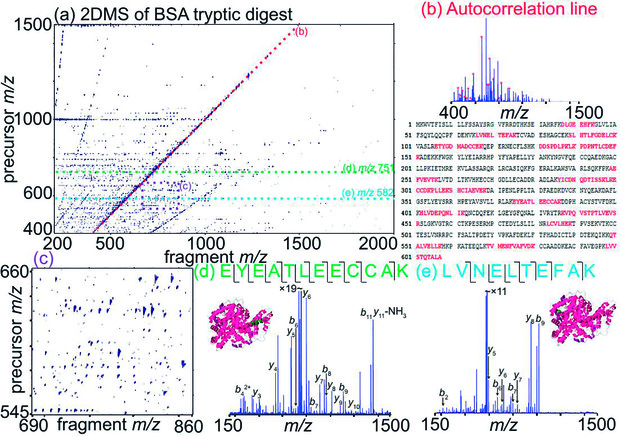
Figure 1 shows the 2D mass spectrum of bovine serum albumin (BSA, 67 kDa). The horizontal axis shows the mass-to-charge ratio of the fragment ions, and the vertical axis shows the mass-to-charge ratio of the precursor (i.e. ionized peptide). Each peak corresponds to a fragmentation. The autocorrelation line (figure 1b), in which the precursor mass-to-charge ratio equals the fragment mass-to-charge ratio, shows the ions from the sample, i.e. the mass spectrum of the tryptic peptides from the BSA protein. Each horizontal line shows the fragmentation pattern of a single precursor ions, which leads to the sequencing of each tryptic peptide (Figures 1d and 1e). Neutral loss lines that are parallel to the autocorrelation line pinpoint the peptides that have post-translational modifications. The zoom-in (figure 1c) shows the 13C isotopic distributions of fragmentations. Using the slope of the line crossing these peaks, we can easily trace back the charge state of the precursor peptide. That way, we can easily correlate the precursor and fragment ions of peptides that have very close mass-to-charge ratios but different charge states. In MS/MS, this demands very stringent isolation conditions.Acquiring 2D mass spectra with different fragmentation methods can therefore give us complementary structural information on proteins and peptides
[caption id="attachment_57468" align="alignnone" width="620"] Figure 1: 2D mass spectrum of a tryptic digest of bovine serum albumin (BSA), with the autocorrelation line, a zoom-in of the spectrum and two fragment ion scans. The tryptic peptides we were able to identify are in red on the protein sequence.[/caption]
Figure 1: 2D mass spectrum of a tryptic digest of bovine serum albumin (BSA), with the autocorrelation line, a zoom-in of the spectrum and two fragment ion scans. The tryptic peptides we were able to identify are in red on the protein sequence.[/caption]
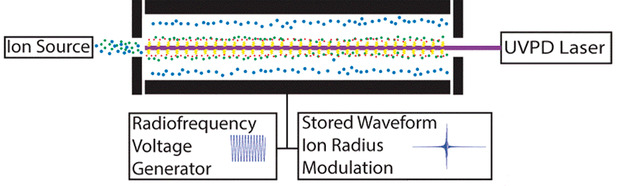
The downside of 2D MS is that an FT-ICR mass spectrometer is a large, expensive instrument with a superconducting magnet. 2D mass spectra can take over an hour to acquire. 2D MS can therefore be coupled with offline preparative LC, but not with online LC. So we proposed a cheaper and faster way of acquiring 2D mass spectra with a linear ion trap. Figure 2 shows how 2D MS in a linear ion trap (LIT) works. The ions are trapped in the LIT with a radiofrequency voltage on the quadrupole electrodes and a continuous trapping voltage on the end-cap electrodes. We used a pulse sequence using stored waveform ion radius modulation (SWIM) that was first proposed by Charles Ross III at Ohio State University for 2D MS in an FT-ICR MS, but which, unlike Gäumann’s pulse sequence, can be transposed to an LIT. We can use laser-based fragmentation methods to generate 2D mass spectra, and we hope to be able to use electron-based fragmentation methods as well.
[caption id="attachment_57469" align="alignnone" width="620"] Figure 2: 2D mass spectrometry with a linear ion trap.[/caption]
Figure 2: 2D mass spectrometry with a linear ion trap.[/caption]
We have validated this method for 2D MS with ion trajectory simulations, and we are now ready to start building a prototype of this device. Linear ion traps are ubiquitous in mass spectrometers. The implementation of 2D MS mainly requires some additional electronics to apply the SWIM pulses to the electrodes and software changes. Linear ion traps can also be used in conjunction with different mass analysers and detectors. In an FT-ICR mass spectrometer, the mass analysis and ion detection takes about 1s per mass spectrum, which is a major contribution to the duration of each 2D MS experiment, which requires 2048 mass spectra. If a linear ion trap is coupled with a time-of-flight analyser, however, the mass analysis and detection only takes 10 ms. Furthermore, the fragmentation method we have used for 2D MS so far (IR laser or electron gun) can take at least 100 ms per fragmentation step.With an ultraviolet laser, this step can be reduced to 10 ns and yield more comprehensive sequencing for peptides.
In this configuration, with 2D MS being applied to a linear ion trap coupled with a time-of-flight analyser and UV fragmentation, the duration of the experiment can be reduced from 1 hour to 10s, which makes 2D MS compatible on an LC timescale. We expect that this will increase the structural information available in each LC-2D MS experiment by over three orders of magnitude. We hope that LC-2D MS will lead to more in-depth biochemical data to study the interactions of biomolecules with cancer drugs for more targeted treatments, among others.Linear ion traps can also be used in conjunction with different mass analysers and detectors

Author: Dr Maria van Agthoven is a Research Fellow in the Department of Chemistry at the University of Warwick
Further Reading
1. Pfändler, P. et al. (1987) Two-dimensional Fourier transform ion cyclotron resonance mass spectrometry. Chem. Phys. Letts. 138: 195-200. 2. Ross, C. et al. (1993) Two-dimensional Fourier transform ion cyclotron resonance mass spectrometry/mass spectrometry with stored-waveform ion radius modulation. J. Am. Soc. Chem. 115: 7854-7861. 3. van Agthoven, M. et al. (2013) Towards analytically useful two-dimensional Fourier transform ion cyclotron resonance mass spectrometry. Anal. Bioanal. Chem. 405: 51-61. 4. Chiron, L. et al. (2014) Efficient denoising algorithms for large experimental datasets and their applications in Fourier transform ion cyclotron resonance mass spectrometry. Proc. Nat. Acad. Sci. 111: 1385-1390. 5. Chiron, L. et al. (2016) SPIKE a Processing Software dedicated to Fourier Spectroscopies. https://arxiv.org/abs/1608.06777. 6. Simon, H. et al. (2016) Uncoiling collagen: a multidimensional mass spectrometry study. Analyst 141: 157-165. 7. van Agthoven, M. et al. (2016) Two-Dimensional Mass Spectrometry for Proteomics, a Comparative Study with Cytochrome c. Anal. Chem. 88: 4409-4417. 8. Floris, F. et al. (2016) 2D FT-ICR MS of Calmodulin: a top-down and bottom-up approach. J. Am. Soc. Mass Spectrom. 27: 1531-1538.
