Overcoming the barrier to ALS
9 May 2013 by qwbdchbctt.d qwbdchbctt.d



Amyotrophic lateral sclerosis (ALS) is a neurodegenerative condition for which there is currently no effective treatment. Here we highlight the importance of enhancing drug delivery to the brain of potential treatments of this devastating disease
Amyotrophic lateral sclerosis (ALS) – also referred to as Lou Gehrig’s disease, Charcot disease, or motor neuron disease – is a neurodegenerative disease characterised by rapidly progressive weakness, muscle atrophy and spasticity, difficulty speaking (dysarthria), difficulty swallowing (dysphagia) and difficulty breathing (dyspnea). The disease occurs relatively uniformly in Western countries, with an average incidence of 1.89 per 100,000 population/year and an average prevalence of 5.2 per 100,000 people1. The mean age of onset for sporadic ALS is about 60 years2,3 and there is a slight male prevalence with a male:female ratio of ±1.5:11. Only 5% of cases have an onset before the age of 30, although juvenile sporadic onset cases are being increasingly recognised4. ALS is the most common adult motor neuron disease, yet the pathogenesis has not been completely unraveled. It is likely to be a complex interplay between multiple pathogenic cellular mechanisms, operating either singly or in combination5,6.
In around 90% of the cases there is no family history of the disease (sporadic ALS, sALS) and therefore the cause for ALS remains unknown7. Several potential causes have been investigated, from toxic influences to free radical damage to nutritional deficiencies, but without consistent findings. The remaining 10% of patients have a known hereditary factor (familial ALS, fALS). The most common mutation is a mutation in the Copper-Zinc superoxide dismutase (SOD1) gene. Transgenic mice overexpressing mutant superoxide dismutase-1 (SOD1G93A) display neuronal cell death in a manner that is consistent with the profile of cell death seen in human ALS patients, and can thus be used as a model for the condition8.
Although ALS is considered incurable, many of the symptoms are treatable, which supports efforts to improve quality of life for as long as possible9. Up to this moment, Riluzole (Rilutek, Sanofi-Aventis) is the only FDA approved drug on the market, however, it only has a modest effect on prolonging life in ALS patients (for up to 2-3 months when taken for a 18-month duration)10-14. Riluzole is generally well tolerated, but has common side effects like asthenia, nausea, gastrointestinal upset and abnormal liver function.
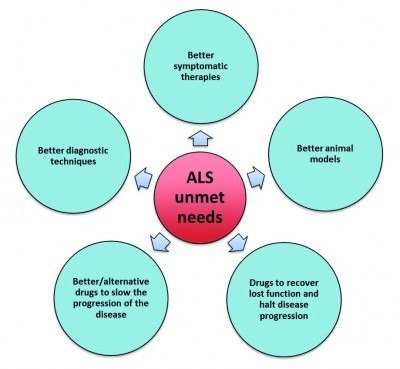
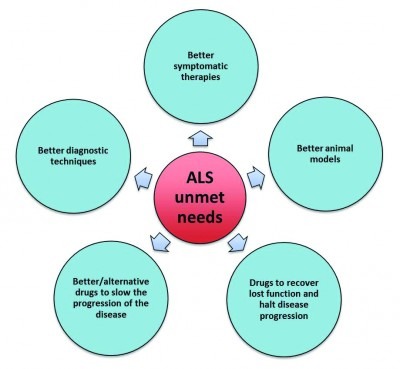
Currently, there are 209 studies in the clinical trial register of the U.S. National Institute of Health. This shows the many attempts to find a cure for this devastating disease. Some of these attempts go beyond the testing of ‘classical’ drug molecules, for example by monitoring the effect of exercise and dietary recommendations, and more invasive such as stem cell transplantations. Not only pharmaceutical or biotechnology companies have an interest in finding new treatments; many patient organisations and research foundations, like the US-based ALS Association and ALS Therapy Development Institute, the UK-based motor neuron disease association and the international alliance of ALS/MND associations are all focusing on patient care, including finding new treatments in a not-for-profit format (Figure 1).
Looking at the classical drug treatments for ALS, edaravone is currently the most advanced option in clinical research. Edaravone was approved for acute ischaemic stroke in 2001, and an open-label phase II study demonstrated that edaravone reduced oxidative stress in ALS patients and delayed the progression of functional motor disturbances16. The results from an ongoing phase III study with edaravone are expected in 2015.
Another candidate for ALS treatment is a component of traditional Chinese medicines, Ursodeoxycholic acid (UDCA). UDCA has quite recently been subjected to efficacy and safety trials for the disease and results have indicated that is it tolerable in ALS patients, and there is evidence of efficacy.
Unfortunately, several other drugs in development for ALS have recently failed in clinical practice. The largest ever phase III trial in ALS (called EMPOWER, including more than 900 patients) with dexpramipexole failed to meet the primary outcome, a joint rank analysis of function and survival called the Combined Assessment of Function and Survival. Other endpoints, including functional decline, survival, or respiratory decline, and subgroup analyses also failed to show efficacy with treatment, overall or for any subpopulation19. Likewise, in 2011, olesoxime was discontinued, as it did not show a significant increase in survival versus placebo in ALS patients receiving riluzole20. These failures, as well as other failures, were obviously very disappointing for the ALS community, including patients, physicians and researchers.
Overall, it was noted that failed results in clinical research raise key questions about enrollment, the number of people with ALS needed to participate and study duration, particularly to inform and empower phase II go/no go decisions21. A phase III clinical trial with minocycline showed a harmful effect on ALS disease progression22, while preclinical studies showed a survival benefit of 10-22%23,24. The initial phase I/II feasibility studies were not designed to evaluate efficacy, but did show an acceptable safety profile25. Discussing the possible reasons for yet another failure, the dose level of minocycline, the possible interaction with riluzole, the clinical trial design and end-point selection were mentioned22,26. In addition, preclinical research results should be scrutinised before moving into the clinical research phase at all. The failure of lithium in a phase II/III study proved an example of this. A pilot study in mice and humans showed positive effects of lithium on slowing the progression of ALS27. However, several follow-up clinical studies have failed to show this effect28-30. A thorough study by researchers from ALS TDI, who were examining the use of lithium as a positive control, showed that false-positive effects in the SOD1G93A mouse model can be avoided by using a rigorous survival study design, including sibling matching, gender balancing, investigator blinding and transgene copy number verification31.
In the past 10 -15 years, research into the pathophysiology of ALS has pointed towards a prominent role of neuroinflammation. Both in patients (in vivo and post mortem) as well as in the mouse models of ALS, activation or proliferation of microglia and astrocytes were observed7,32-34. This has led to the investigation and development of treatments against neuroinflammatory components. NP001 (Neuraltus), a drug that is designed to restore the normal functioning of activated macrophages, is an example of an anti-inflammatory ALS drug. The recent phase II clinical study demonstrated a positive trend in slowing the rate of ALS disease progression and a phase III study will be started mid 201335. Another example for a neuroinflammatory target is the preclinical investigation into an anti-CD40L antibody. ALS TDI, Biogen Idec and UCB Pharma SA have entered into a collaboration to investigate the use of an anti-CD40L antibody as potential treatment for ALS36.
[caption id="attachment_32997" align="alignleft" width="400" caption="Figure 1: Key unmet needs in ALS treatment. Many factors are unknown in ALS pathogenesis and no biomarker has been identified to diagnose patients in an early stage of the disease. This prevents interference in the earliest stages and lowers survival chances. In addition, ALS patients are in need for better drugs with higher efficacy and disease-modifying effects. The main goal for companies, foundations and patient organizations is to slow the progression of disease or even halt the disease progression, instead of symptomatic treatment. Adapted from Datamonitor15."] [/caption]
[/caption]
to-BBB has recently developed glutathione pegylated liposomal methylprednisolone (2B3-201) for the treatment of neuroinflammation. Proof-of-concept results obtained in a model of Multiple Sclerosis (MS) showed that 2B3-201 was more effective in reducing clinical signs compared to free methylprednisolone and non-targeted pegylated liposomal methylprednisolone37. Together with scientists from the Departments of Pharmacology and Clinical Neurology from the University of Oxford, to-BBB has tested 2B3-201 in the SOD1G93A model. Compared to free methylprednisolone treatment, 2B3-201 treatment resulted in more normal motor neurons, less astrocytosis and resulted in improved neuroprotection. Researchers at the ALS TDI have investigated the pharmacokinetics of brain and spinal cord uptake of 2B3-201 and, based on the positive results that were obtained so far, have recently started a long-term survival study in the SODG93A model. Based on these initial promising results of to-BBB’s G-Technology in the application towards ALS, ALS TDI and to-BBB agreed to investigate additional promising compounds in a joint effort38.
In addition to 2B3-201, to-BBB is working on several other co
mpounds that could have a benefit for patients suffering from ALS. Since we believe that combined efforts have a synergistic effect, we are actively pursuing additional collaborations, next to the ongoing investigations with researchers from Oxford University (UK) and ALS TDI (USA). We are setting up collaborations with Utrecht University (the Netherlands), as well as special purpose companies founded by ALS patients. Such projects could for example use drugs that are approved elsewhere, but not yet available in Europe, and that can benefit from enhanced delivery to the brain and spinal cord.
to-BBB’s core competency is enhancing drug delivery to the brain, using the G-Technology, a brain-targeted liposomal platform. Through the versatility of the liposomes it is possible to encapsulate a variety of drug molecules39, and while most of the efforts we ar
e currently pursuing are still too early to report, we are keeping an open and collaborative mind. For example, within this network of collaborators, we are not only looking into Western-type medicinal chemistry molecules, but also towards traditional Chinese medicine with a potential beneficial effect on ALS. From the literature, as well as general Internet sources, several such traditional Chinese medicines have been described.
For devastating diseases like ALS, for which currently only moderately effective drugs exist, it is important to find disease-modifying treatment options. Since it is a complex disease, the input of many stakeholders is required. Only by combining all efforts worldwide, a safe and efficacious treatment could hopefully be developed in the shortest time possible.
- Worms PM (2011) The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci 191:3-9.
- Leigh PN (2007) Amyotrophic lateral sclerosis. In Motor Neuron Disorders and related diseases Vol 82. Edited by: Eisen AA, Sham PJ. Amsterdam: Elsevier; pp:249-268. [Aminoff MJ, Boller F, Swaab DF (Series Editor): Handbook of Clinical Neurology]
- Haverkamp LJ, Appel V, Appel SH (1995) Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain 118(Pt 3):707-719.
- Gouveia LO, De Carvalho M (2007) Young-onset sporadic amyotrophic lateral sclerosis: A distinct nosological entity? Amyotroph Lateral Scler 8(6):323-327.
- Shaw PJ (2005) Molecular and cellular pathways of neurodegeneration in motor neuron disease. J Neurol Neurosurg Psychiatry 76:1046-1057.
- Cozzolino M, Ferri A, Carri MT (2008) Amyotrophic lateral sclerosis: from current developments in the laboratory to clinical implications. Antioxid Redox Signal 10:405-443.
- Philips T, Robberecht W (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol 10:253-623.
- Scott S, Kranz JE, Cole J, Lincecum JM, et al. (2008) Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler 9(1):4-15.
- Gordon PH (2011) Amyotrophic Lateral Sclerosis; pathophysiology, diagnosis and management. CNS Drugs 25(1): 1-15.
- Bensimon G, Lacomblez L, Delumeau JC, Bejuit R, Truffinet P, Meininger V (2002) A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol 249:609-615.
- Meininger V, Lacomblez L, Salachas F (2000) What has changed with riluzole? J Neurol 247:19-22.
- Mitchell JD, O'Brien MR, Joshi M (2006) Audit of outcomes in motor neuron disease (MND) patients treated with riluzole. Amyotroph Lateral Scler 7:67-71.
- Guidance on the use of riluzole for the treatment of motor neuron disease [http://www.nice.org.uk/nicemedia/pdf/RILUZOLE_full_guidance.pdf]
- Turner MR, Parton MJ, Leigh PN (2001) Clinical trials in ALS: an overview. Semin Neurol 21:167-175.
- Datamonitor (2007) Pipeline Insight: Orphan diseases in CNS. Part II Amyotrophic Lateral Sclerosis. Reference Code: DMHC2354
- Yoshino H, Kimura A (2006) Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph Lateral Scler 7(4):241-245
- Min JH, Hong YH, Sung JJ, Kim SM, Lee JB, Lee KW (2012) Oral solubilized ursodeoxycholic acid therapy in amyotrophic lateral sclerosis: a randomized corss-over trial. J Korean Med Sci 27(2):200-206
- clinicaltrials.gov NCT00877604
- Medscape Medical News, 4 January 2013; www.medscape.com/viewarticle/777141
- Press Release, Trophos SA, 13 December 2011; www.trophos.com/news/pr20111213.htm
- http://blogs.als.net/post/2012/12/14/ALSMND-2012-Clinical-Trials-Tribulations.aspx.
- Gordon PH, Moore DH, Miller RG, Florence JM, et al. (2007) Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol 6:1045-1053
- Kriz J, Nguyen MD, Julien JP (2002) Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 10(3):268-273
- Van Den Bosch L, Tikin P, Lemmens G, Robberecht W (2002) Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport 13(8):1067-1070
- Gordon PH, Moore DH, Gelina DF, Qualls C, Meister ME, et al. (2004) Placebo-controlled phase I/II studies of minocycline in amyotrophic lateral sclerosis. Neurology 62(10):1845-1847
- Swash M (2007) Learning from failed trials in ALS. Lancet Neurology 6:1034-1035
- Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, et al. (2008) Lithium delays progression of amyotrophic lateral sclerosis. PNAS 105(6):2052-2057
- Aggarwal SP, Zinman L, Simpson E, McKinley J, Jackson KE, et al. (2010) Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a randomised double-blind, placebo-controlled trial. Lancet Neurol 9:481-488
- Chio A, Borghero G, Calvo A, Capasso M, Caponnetto C et al. (2010) Lithium carbonate in amyotrophic lateral sclerosis: lack of efficacy in a dose-finding trial. Neurology 75(7):619-625
- Verstraete E, Veldink JH, Huisman MH, Draak T, Uijtendaal EV, et al. (2012) Lithium lacks effect on survival in amyotrophic lateral sclerosis: a phase IIb randomised sequential trial. J Neurol Neurosurg Psychiatr 83(5):557-564
- Gill A, Kidd J, Vierira F, Thompson K, Perrin S (2009) No benefit from chronic lithium dosing in a sibling-matched, gender-balanced, investigator-blinded trial using a standard mouse model of familial ALS. PloS ONE 4(8): e6489 doi:10.1371/journal.pone.0006489
- Evans MC, Couch Y, Sibson N, Turner MR (2012) Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Moll Cell Neurosci http://dx.doi.org/10.1016/j.mcn.2012.10.008
- Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, et al (2004) Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 15, 601–609
- Anneser JM, Chahli C, Ince PG, Borasio GD, Shaw PJ (2004) Glial proliferation and metabotropic glutamate receptor expression in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 63: 831–840
- Press Release, Neuraltus Pharmaceuticals, 29 October 2012; www.neuraltus.com/pages/news_rel10_30_12.html
- Press Release, ALS TDI, 19 December 2011; www.als.net/Media/5407/News/
- Gaillard PJ, Appeldoorn CCM, Rip J, Dorland R, van der Pol SMA, et al (2012) Enhanced brain delivery of liposomal methylprednisolone improved therapeutic efficacy in a model of neuroinflammation. J Control Release 164(3): 364-369.
- Press Release, ALS TDI and to-BBB, 8 January 2013; www.als.net/Media/5427/News/
- Gaillard PJ, Visser CC, Appeldoorn CCM, Rip J (2012) Enhanced brain drug delivery: safely crossing the blood-brain barrier. Drug Discov Today: Technol 9: 155-160.
Authors: Pieter Gaillard, Corine Visser, Manon de Waard, Marco de Boer, and Chantal Appeldoorn
to-BBB technologies BV, Leiden, the Netherlands
Phone +31 (0)71 3322255
Fax +31 (0)84 8313409